 This page describes a more accurate spatial absorption calculation which minimizes interpolation errors that occur because of discontinuities in the electric field near interfaces. Consider using the advanced method if a significant fraction of the absorption occurs within the first mesh cell at interfaces.
This page describes a more accurate spatial absorption calculation which minimizes interpolation errors that occur because of discontinuities in the electric field near interfaces. Consider using the advanced method if a significant fraction of the absorption occurs within the first mesh cell at interfaces.
Background
This technique is an extension of the simple method described on the previous page. For reasons given on the previous page, the advanced method calculates the absorption profile using the relation
$$ P_{abs} = -0.5 \ \omega \vert E \vert ^2\ imag(\varepsilon)$$
The difference between the two techniques is the monitor interpolation option.
An inherent part of the FDTD method is that during the simulation, each field component (Ex, Ey, Ez) is calculated at a different location within the Yee cell. If a monitor collects raw field data from the simulation, post processing becomes complicated due to the fact that each field component is known at a different location. Even simple calculations like |E|^2 = |Ex|^2+ |Ey|^2+ |Ez|^2 are not trivial since Ex, Ey, and Ez are not known at the same location.
To simplify post-processing of simulation data, the default behavior of monitors in FDTD is to interpolate all field components back to a common set of points (the origin of the Yee cell). This makes post-processing simple, since all field components are known at a common point.
In most situations, the interpolation errors that occur when interpolating each field component back to the origin of the Yee cell is negligible. Unfortunately, the absorption calculation can be quite sensitive to these interpolation errors. The interpolation errors only become significant when:
- The electric field component normal to the structure surface is large AND
- a significant fraction of the total absorption occurs within the first set of mesh cells at the structure surface.
From Maxwell's equations, we know that the field component normal to the structure surface will be discontinuous at the interface. Also, it is obvious that interpolating near a discontinuity can lead to errors. In the absorption calculations, the interpolation error typically makes the electric field just inside the absorbing material much larger than it really is. Since absorption is proportional to electric field intensity, this will tend to overestimate the total absorption.
Note that these errors only occur for the mesh cells which cross the material interface. If the absorption in the mesh cells that cross the interface is a small fraction of the total absorption, then the overall error will be small. However, if most of the absorption is concentrated near the surface (as is the case with metals), then the overall error can be significant.
|
Note: Conformal mesh The material data has an index that is on the same order of magnitude as the background index. Also, we are able to simulate this problem at a reasonably small mesh size of 5nm or less. For this reason, we can change the mesh refinement option to "conformal variant 1" to take full advantage of the conformal meshing feature, even for a metals. |
Example
The simulation file usr_absorption_advanced.fsp can be used to calculate the power absorption per unit volume of a gold sphere illuminated by a focused beam.
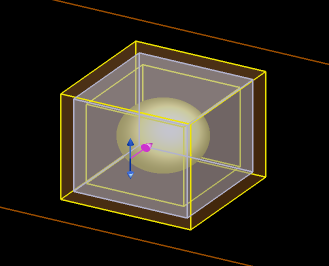
The total absorption is calculated in three ways:
- A box of 2D monitors around the sphere calculates the net power absorbed within the box. This is an easy, efficient and accurate technique to calculate total absorption within a rectangular region.
- The simple absorption analysis group described in the previous page. The monitors in this group use the standard field interpolation option. It will not give accurate results in this simulation because the field component normal to the surface of the gold sphere is large. Errors in the absorption data near the surface are significant since most of the absorption occurs near the surface.
- The advanced absorption analysis group. The monitors in this group do not use monitor interpolation. This gives more accurate absorption data near the sphere surface.
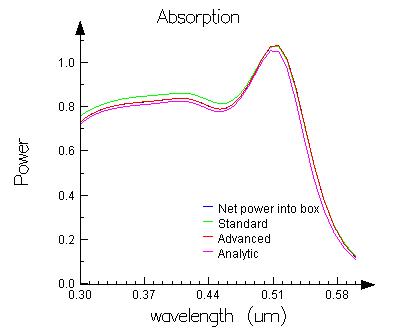
Run the simulation, then run the associated analysis script. It will create a figure showing the absorption spectrum as calculated by the three techniques. The analytic solution (calculated from Mie theory) is also shown.

There are a few interesting points to notice:
- The Net power into box and Advanced techniques are very similar.
- The absorption calculated by the standard technique is quite different than the result from the other techniques The standard technique is not appropriate for calculating the spatial absorption profile in metals because it is very sensitive to interpolation errors of discontinuous field components at the metallic interfaces.
- All three techniques are somewhat different than the analytic result. This example was setup to run quickly, and not to provide the most accurate result. A smaller mesh size, larger simulation region, and more PML layers would be required to get closer agreement with the theory.
|
Tip: Size of the monitor To get the most accurate results, the analysis object should be large enough to completely surround the absorbing material, plus at least 1 mesh cell of non-absorbing region around the edges of the analysis object |
|
Tip: Periodic and Bloch boundary conditions To get the most accurate results when using this analysis object with Periodic and Bloch boundary conditions, a small change should be made within the Analysis script. For details, see the analysis script comments or contact Lumerical technical support. |
|
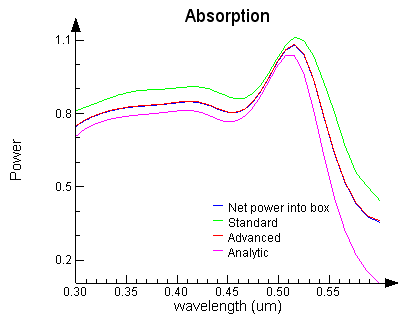
Note: Higher accuracy simulation The following figure shows the absorption data from a simulation that was run with higher accuracy settings. As you can see, the FDTD results are converging to the analytic solution. The simple Net power box and Advanced technique are closest to the theory.
To increase the accuracy of the example file, make the following changes:
Warning: The memory requirements and file size will be very large with these changes. |